This blog discusses genome foundation models. The first part of the blog provides more of an explanation for the concepts of a foundation model and is designed to introduce the topic. The second part of the blog shares some opinions on current protein and genome language models.
What is a Foundation Model
A foundation model learns a broad set of knowledge that is useful in an array of various tasks. The idea is that developers can build on top of that model to solve problems better and more quickly. In addition, a foundation model may itself be directly capable of tasks, and so users may directly use the foundation model as a user.
The ability of a foundation model to serve either purpose is a function of the information learned by the model. What is learned by a model is an interaction between:
- The architecture and training machinery of the model
- The data that is shown to the model during training
- The task that the training process requires the model to solve.
As a model is trained to solve a task, the process of training organizes the information in the network, and encodes knowledge that is required to solve the task effectively.
Walking through an example task
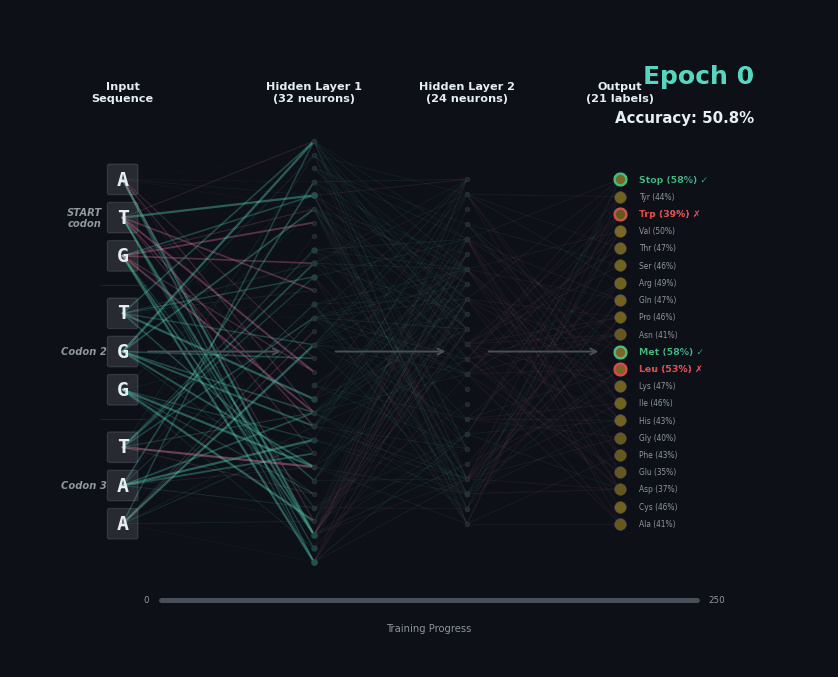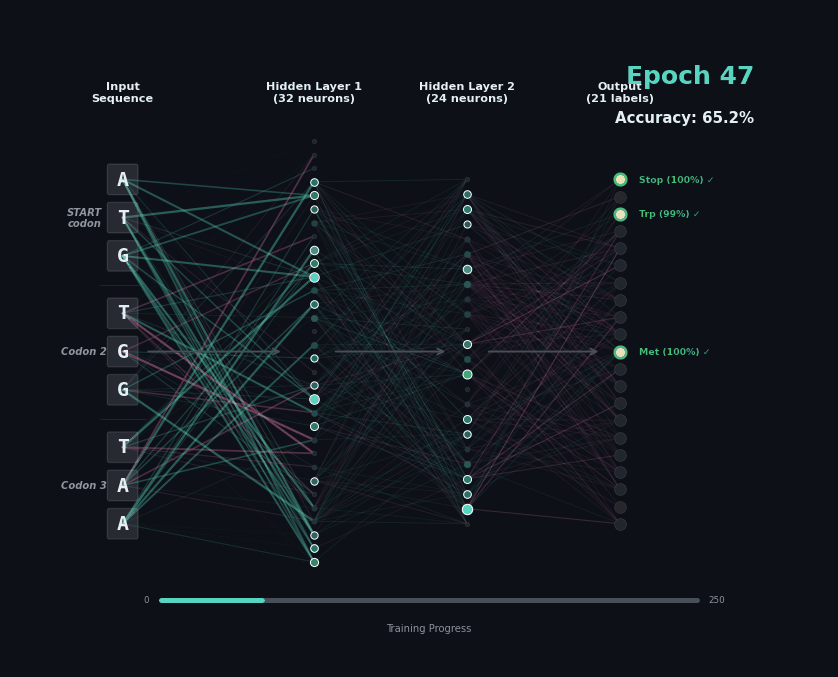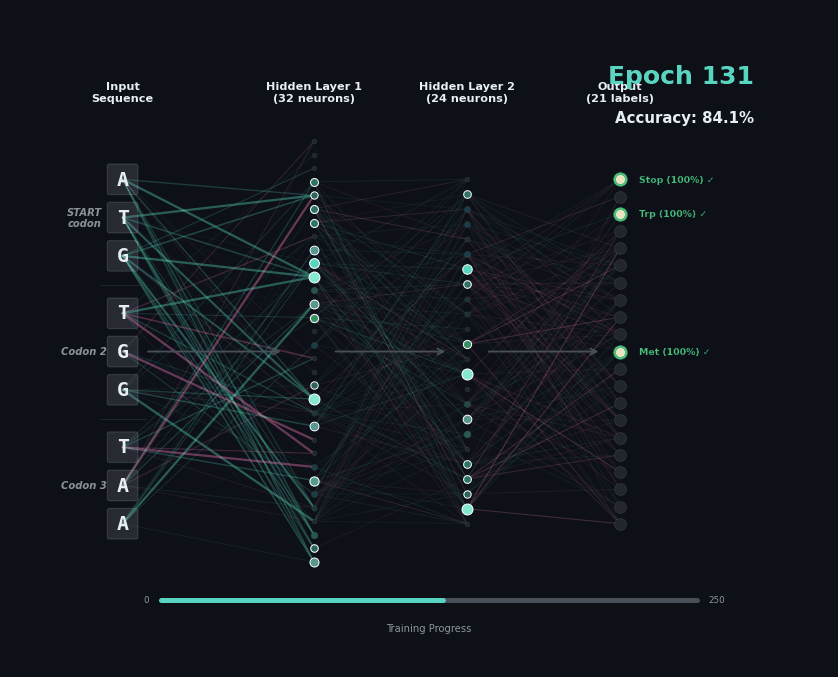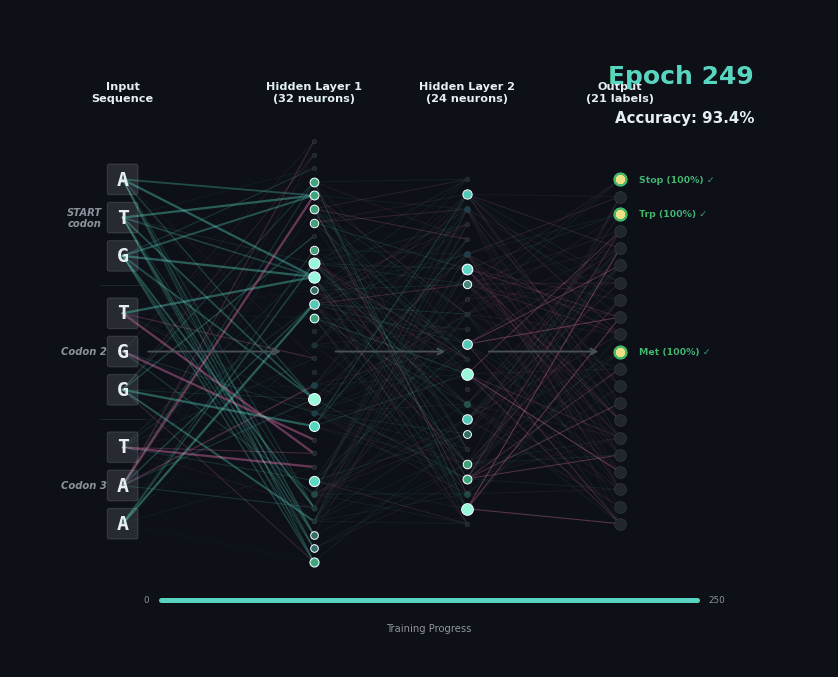
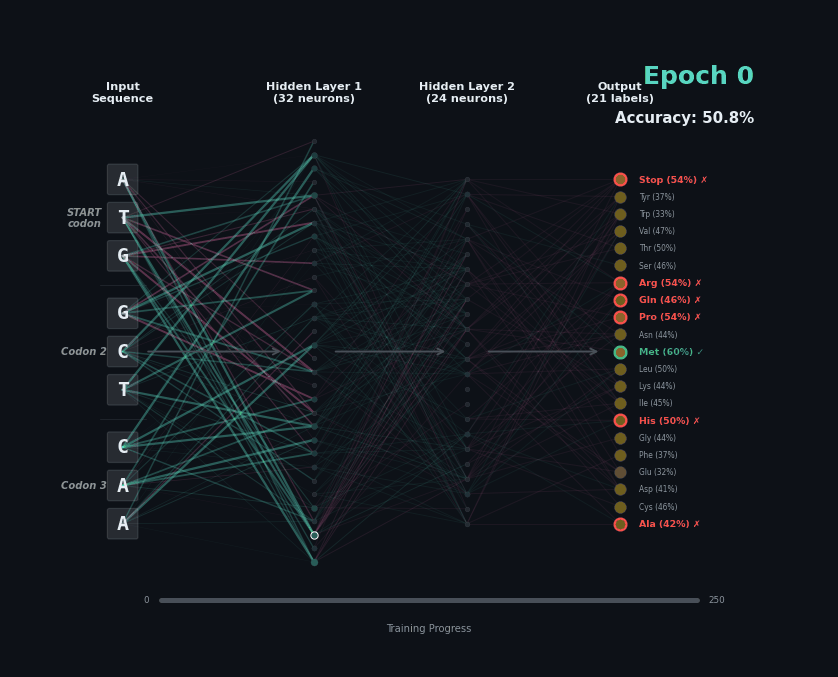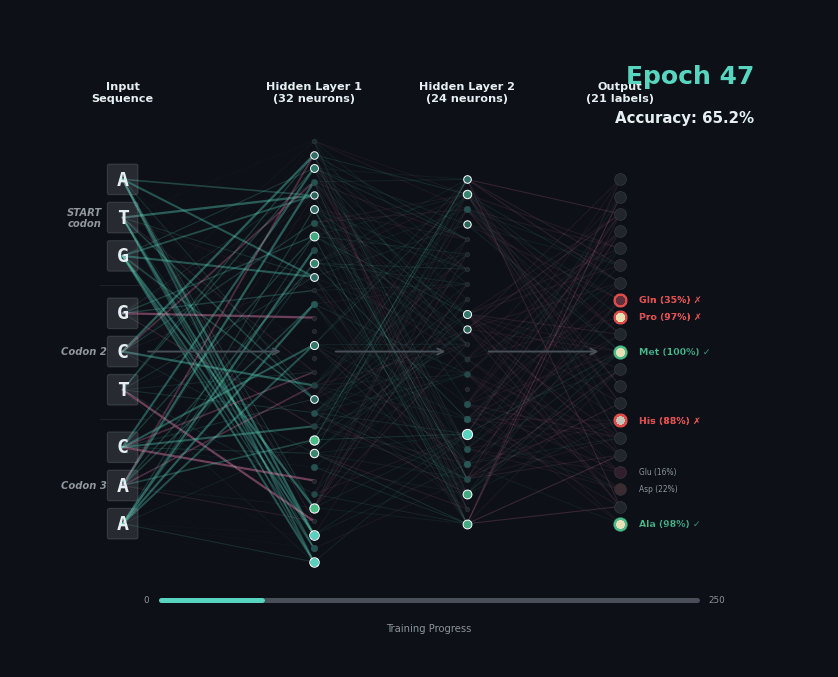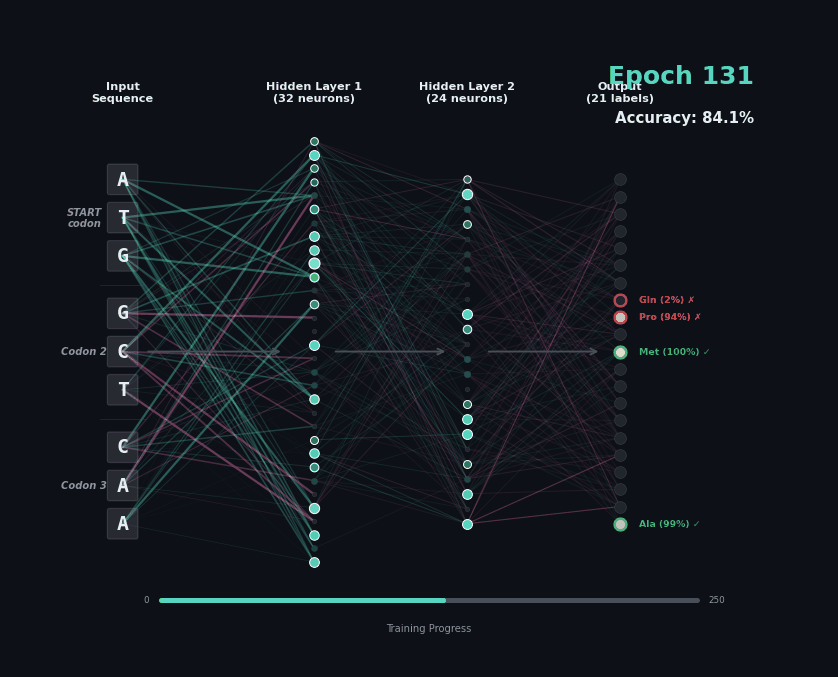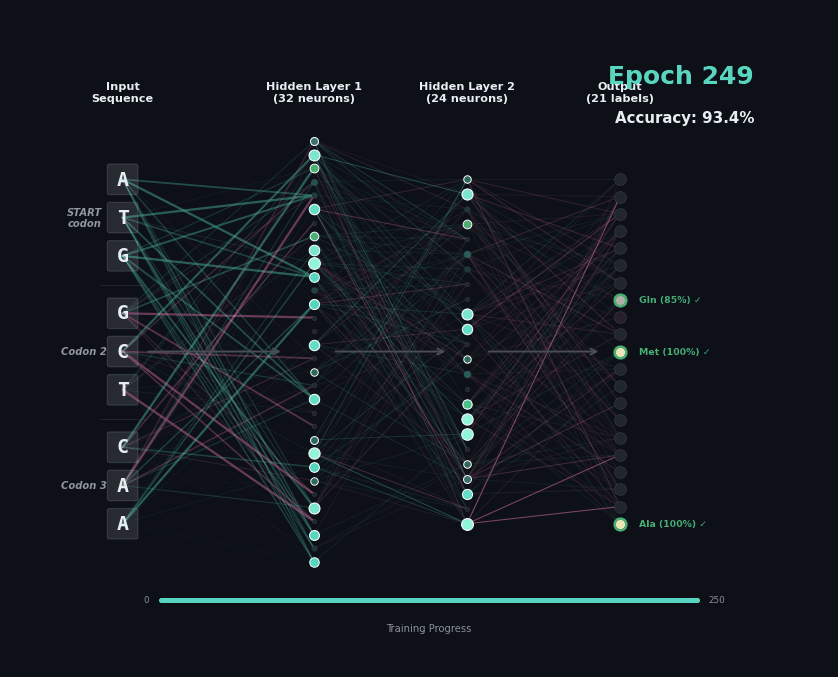
Let’s train a model with the following task. Given a set of 9 nucleotides, report the amino acids encoded by the reading frame. E.g. if given ATGAGGGTT, report MRV, if given TTGAGGGTT, report nothing as the first codon is not start. To solve this task, the model must learn the rules of the genetic code.
I’ve trained a simple 3-layer network to solve this problem. Training models like this involves showing a series of labelled examples and updating the weights of the model based on whether a prediction is correct. Training progresses by showing the data in an “epoch” and over training the model progressively learns how to predict examples.
Below I am showing how two examples, (MW-STOP) and (MAQ) are predicted over the course of the training run. I am showing the activation of neurons on their connection, as well as the training epoch and the overall accuracy on the hold-out test set of the model.
 |
 |
Transferring knowledge from a foundation model to a new model
In order to solve this task, our model has to learn rules of translation - how the triplet code corresponds to amino acids. Let’s see how this information can be useful to help solve different, but related tasks.
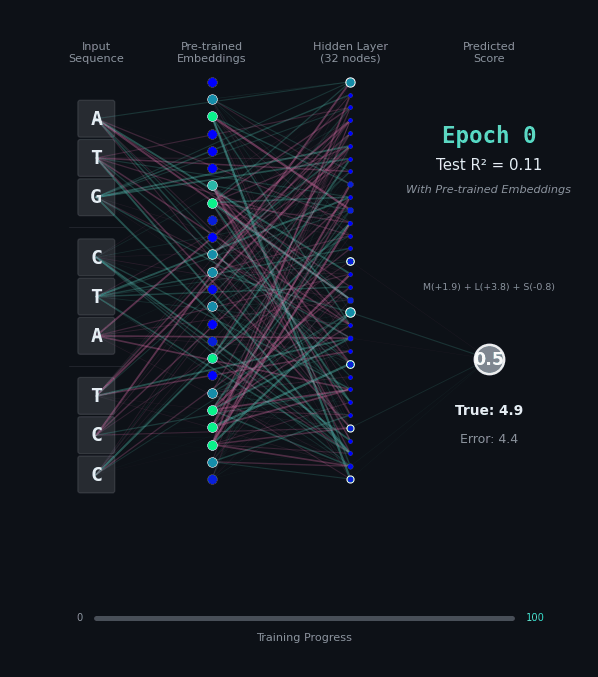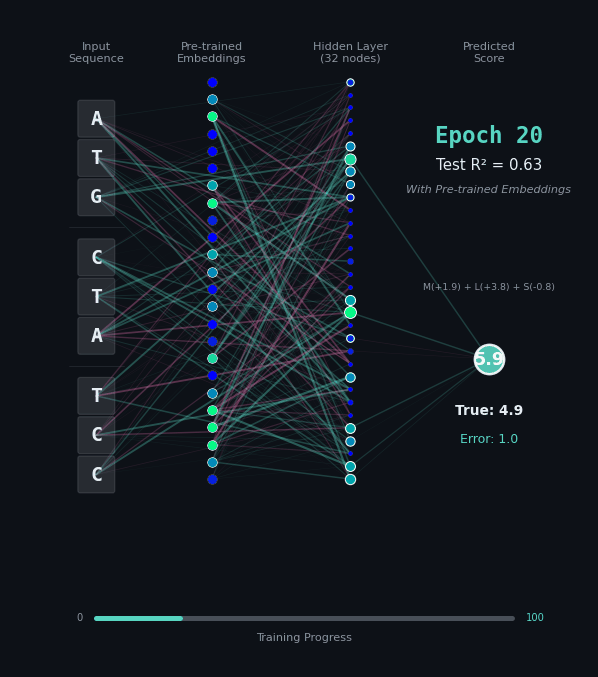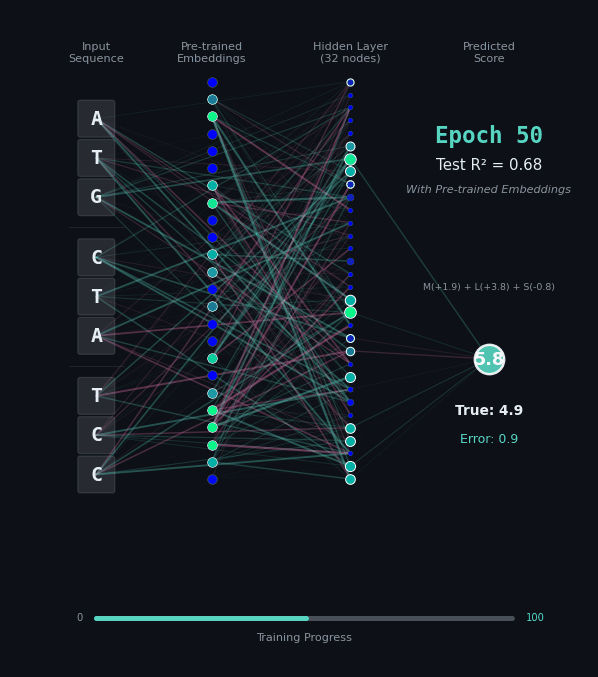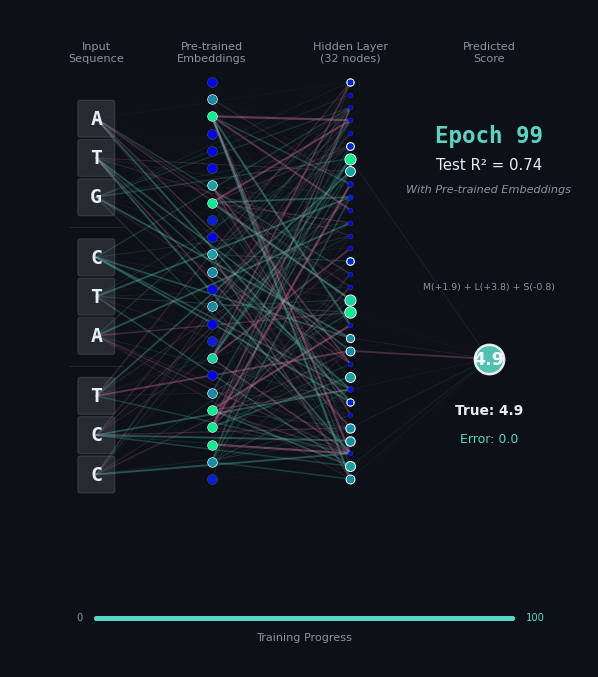
For this task, let’s predict the hydrophobicity score of the amino acid sequence, by summing up the hydrophobicity of each amino acid encoded (if any). So for the amino acids M (+1.9), L (+3.8), S (-0.8) we should predict 4.9. Solving this task is a bit harder, because we still have to solve the codon rules, and then we have to map the amino acids onto their properties.
Here, I’ll show you two different networks. In the first network on the left, we are going to train the model “from scratch” - meaning directly on the nucleotide data. In the second model on the right, we are going to take the connections learned in the network from the first task as a layer of pre-trained embeddings. The concept of embeddings is important, embeddings represent either directly or in a compressed form the information learned in some part of the network. Our new network is going to learn from that network how to classify the task.
Like before, I’ll show you the network for two examples. Notice how the model on the right with the embeddings learns to solve the problem faster and with an overall better accuracy.
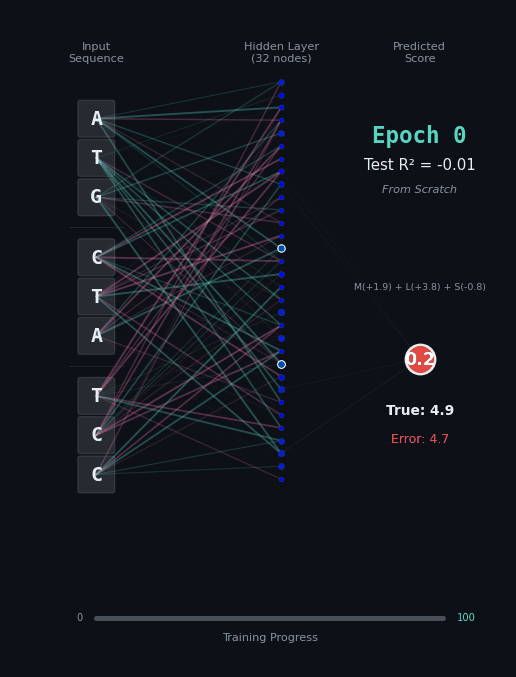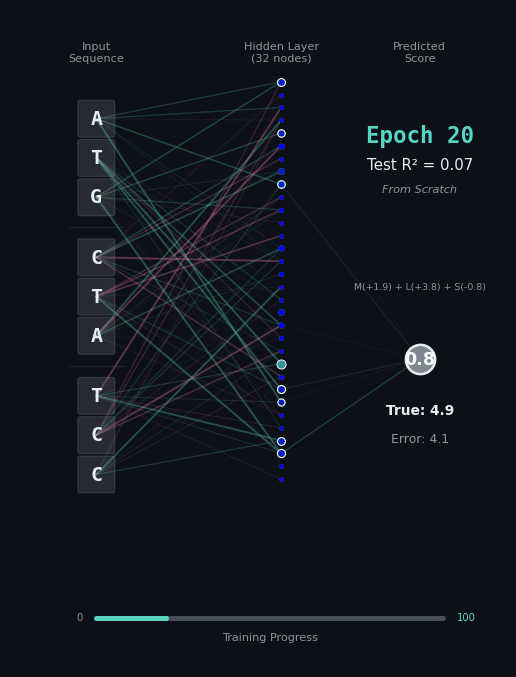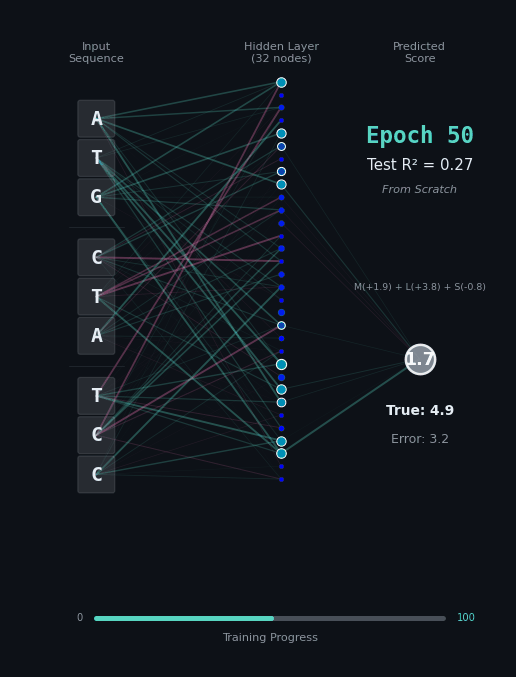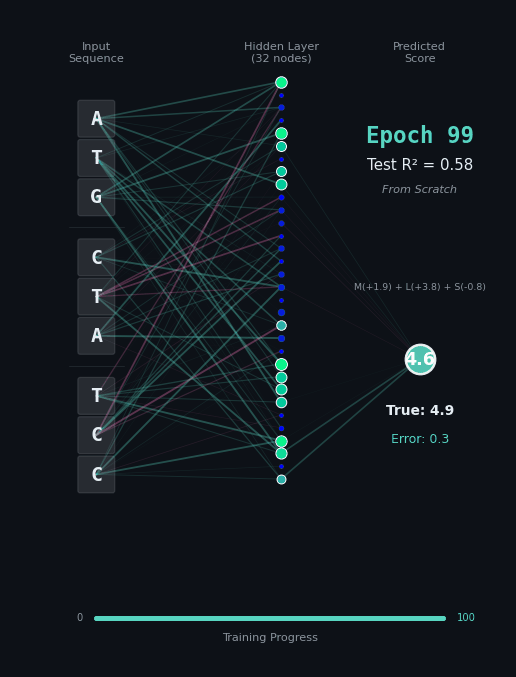 |
 |
What are the advantages of the foundation model approach
This approach is most useful when your ability to encode knowledge into the network is limited compared to what was used to train the foundation model. Since that knowledge is a function of training data and task, here are the most common places that would be the case:
- When you are solving a more narrow task, and although there is useful general knowledge in training for diverse tasks, you prefer to focus your training setup on the task you care about.
- When you are data limited. You may have a handful of problems in our domain, and are unable to train a model with broad knowledge.
- When you are compute constrained, and want to use your resources more efficiently.
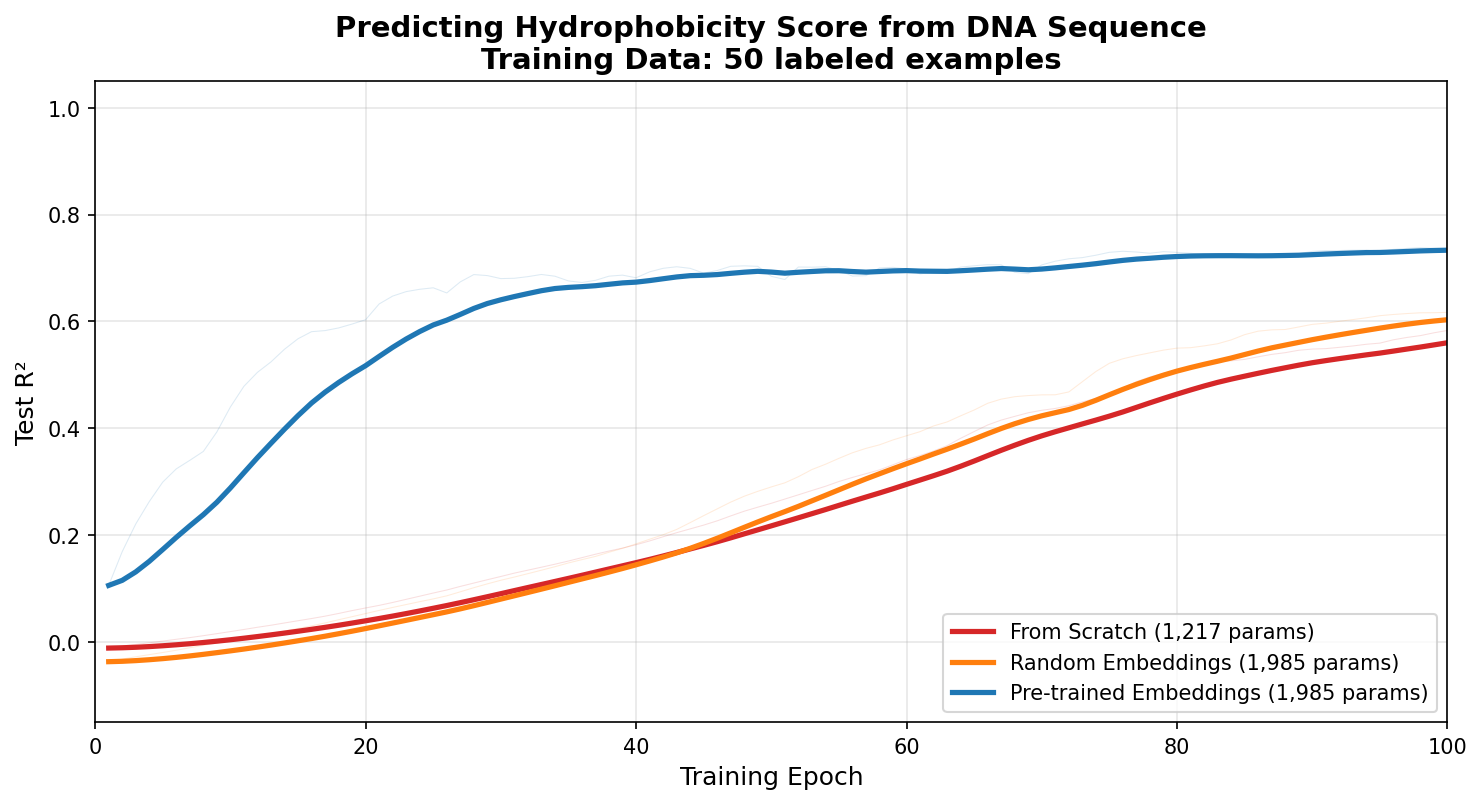
To illustrate these advantages, we can look at some training curves. The curve below shows the evaluation accuracy in training a model on 50 examples. The blue curve is model with pre-trained embeddings. You can see how it can rapidly focus learning on the hydrophobicity problem, while the from scratch model in red has to learn both aspects of the problem. To show the importance of the prior knowledge, in the orange curve I took the same architecture of the model with embeddings, but randomized the weights, basically erasing the information in the network. This model has the same architecture as the embeddings model, but without the prior information does not give the performance boost.
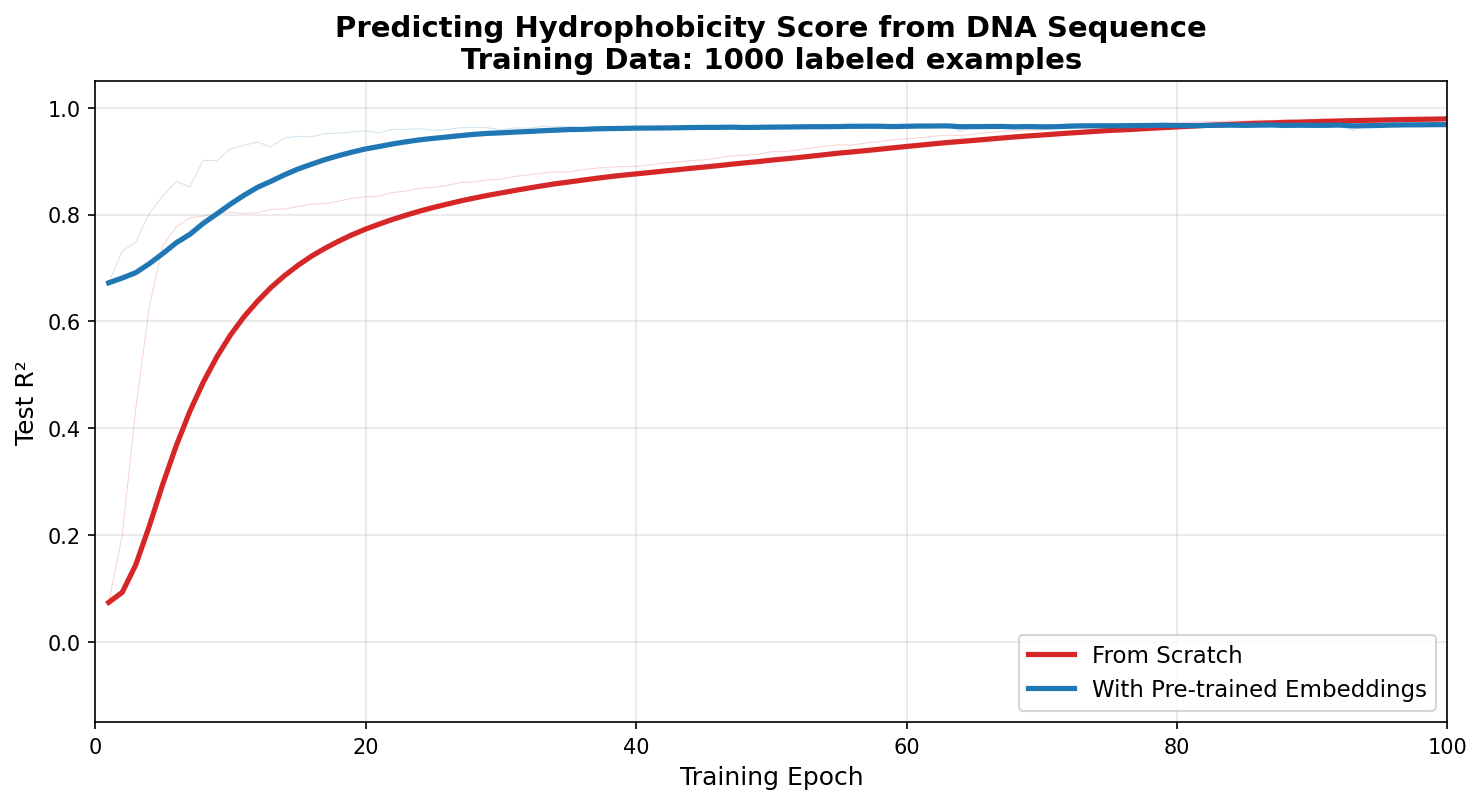
To illustrate how the amount of data affects the relative advantage of using foundation models, in this example I am expanding the training dataset from 50 examples to 1,000. Here, you can see that the model with the embeddings still learns much faster, but with the larger amount of data, the “from scratch” model can eventually reach the same performance.
Inspecting what models learn
To emphasize again - the information in a model is shaped by the tasks it needs to solve in training, and the effectiveness of the training data and setup that let it solve the tasks.
One of the major advantages to using the information in neural networks is that they allow data points to be compared within a semantic space that is organized by the tasks. So while comparing nucleotide sequences as kmers would space examples similarly apart by match or mismatch, comparing the information in these embeddings can give a nuanced view of their similarity.
To make this point, let’s visualize the information in these two networks. Here, I have taken a set of examples that repeat each amino acid (MAA, MCC, MDD, … , MWW) and shown the nucleotide sequence to the codon model from task 1 and the hydrophobicity model from task 2.
For each example, the neurons in the middle layers will activate in different ways. I have taken the vector for each of the activations and used PCA to do a dimensional reduction so we can see the similarity in how the neural network responds to the different inputs.
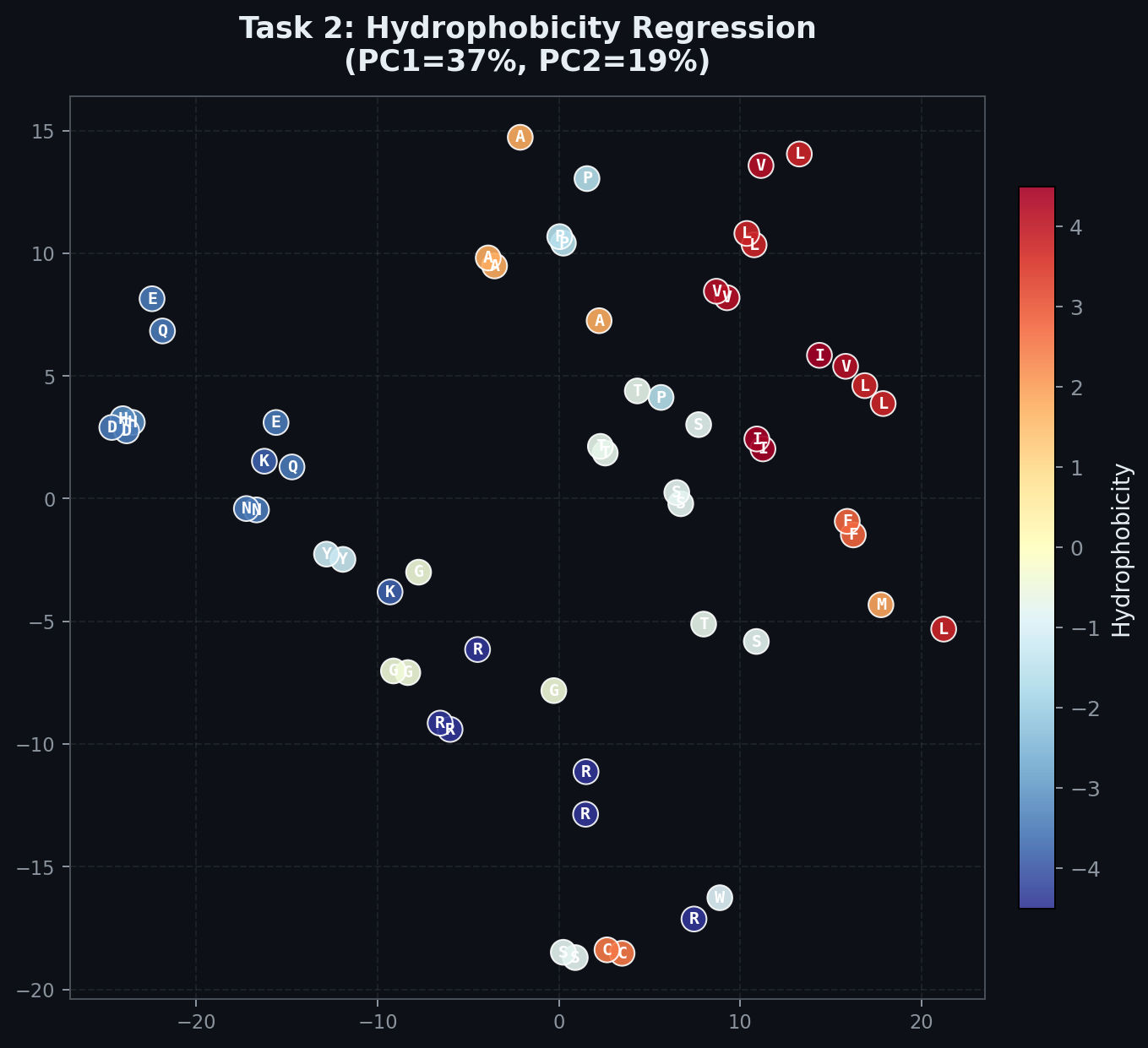
This is the PCA for task 2 (hydrophobicity prediction). You can visually see the separation in neuron activation for amino acids that are more hydrophobic versus less (the repeated characters are for amino acids with multiple ways to encode them e.g. TGT, TGC for Cysteine).
Interestingly, you may be able to see some artifacts of the original embedding, here cysteine (C), a hydrophobic amino acid, is close to Tryptophan (W), a neutral one. Cysteine and Tryptophan share their first 2 nucleotides (TG) with STOP.
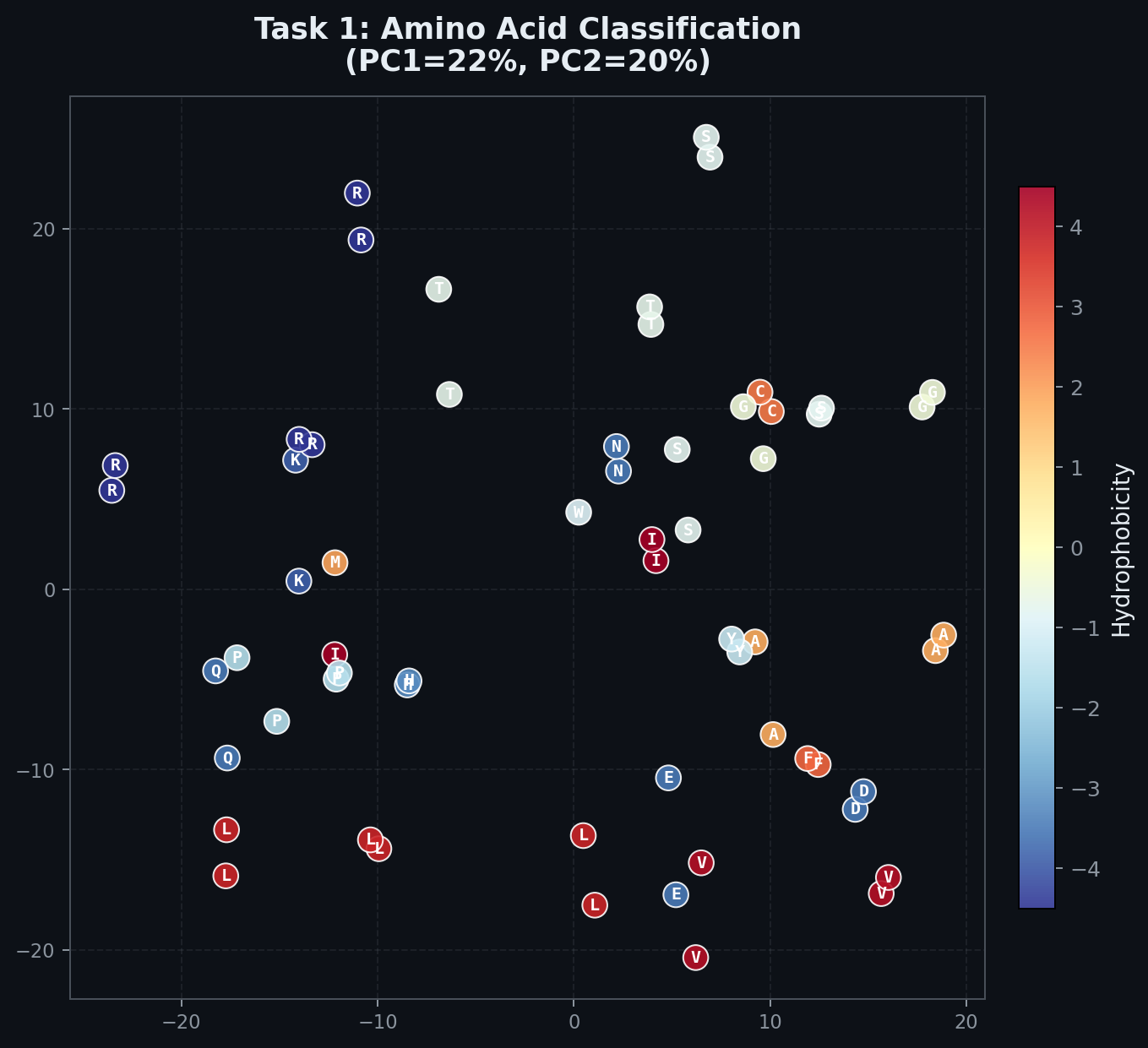
In contrast, this is the same PCA for the network in Task 1. You can see that there is no clear structure in terms of hydrophobicity. Also interesting to reflect on is the fact that we actually do expect to see some structure, because of the design inherent in the triplet code itself. The triplet code is organized in a manner that places similar amino acids closer to each other, in order to minimize the disruptive effect of a mutation. To the extent that we see structure in the image, it is because the shadow of that structure is implicit in the original task.
What’s the difference between any model and a “foundation model”
The models we build here are very simple. Although no one would consider them foundation models, we can use them to build other models with the same principles. In this sense, any model can be used as a foundation, and there is a rich set of transfer learning methods for how to do this.
When the term “foundation model” was first coined, this annoyed me to some degree, because it wasn’t clear to me that a new term was necessary. Subsequently, I’ve come to feel that the term can be useful - the defining feature of a foundation model is its breadth. Specifically, it’s designed to be useful to a diverse array of tasks which you would usually build specific models for. It may not solve all of those tasks as well as alternative solutions, it may not even solve any single task better than an alternative, but it will be broadly applicable.
Thoughts on Genome and Protein Language Models
I’m going to transition to share thoughts on genome language models, such as Evo/Evo2 or HyenaDNA, and protein models such as ESM2 or AlphaFold (though here I am thinking more of ESM2 as it is a more purely sequence based model. This section is more my opinions.
Protein models such as ESM2 are generally:
- Directly useful, often able to outperform alternative, specialized methods
- Their embeddings are often directly useful - e.g. if you want to add “information from a protein’s sequence” alongside other parts of a model
In contrast, Genome (nucleotide) models generally:
- Only conditionally outperform alternative, specialized models. Most tasks with large amounts of data/examples favor specialists
- Their embeddings are useful to accelerate training on tasks that operate on nucleotide data, and can lead to better performance in data limited cases or in needle-in-haystack problems.
So generally, protein models are more likely to be useful out of the box, and so benefit users directly as well as developers. Whereas genome models are currently most useful to developers.
I think that for genome models, the difference in usefulness to different types of people results in a mixed opinion throughout the community. People who want a directly useful tool quickly find fault, while there is another group benchmarking models to identify whether the models can improve training for their tasks.
Thoughts on why it’s harder to make Genome Language Models than Protein Ones
There are many brilliant people working on both genome and protein language models. The difference in outcome isn’t from talent or effort, I think it’s an expected consequence of the data and problem the models solve.
The most common way to train language models (general text, DNA, or protein) is to blind the model to certain tokens and ask the model to reconstruct the original sequence from partial information.
Human language is very information dense. Every word has conscious intentionality behind its selection. Even in cases of similar words (joy vs. elation vs. rapture or compel vs. coerce vs. oblige), there are subtle shades which reinforce the intention of the writer, how the reader is meant to hear, or the meaning of what is communicated. The tokens in human writing, especially good writing, are incredibly information rich.
Similarly, the vocabulary of amino acids each correspond to physical properties, (e.g polar, bulky, flexible). The world of protein language is more closely linked to the physical world.
DNA can be less constrained. Long stretches of it can have no or nearly no reason for one base being present versus another. Parts important to understand an other part can be located very far away (for example a distal enhancer). This is one reason why some of the most interesting innovations in DNA language models are in extracting information in extremely long contexts.
Although it is true that the DNA model encodes proteins (though due to the triplet code, with more degeneracy), the same model which has to learn the coding portions has to learn the general parts as well. Because training moves the model globally away from reconstruction error, regardless of where it comes from, the effects of model training are spread across both information dense and information sparse tasks. You may comment then that we could instead train on prokaryote genomes which are more streamlined, but then these genomes are much shorter and the training data is less than the eukaryote genomes.
I think that DNA language models are useful now, as tools to accelerate or improve training for data or compute limited tasks. I am also optimistic that there will be more powerful genome language models in the future. I think some of the things that will lead to better models are converting the training task into things which are more information dense (e.g. predicting the distribution of nucleotides in an alignment of genomes so you can predict unconstraint as a meaningful prediction). Also, training more directly on multiple tasks or questions you can directly ask of nucleotide sequence directly in pre-training. In short, things which are able to concentrate the meaningful information density significantly at the level of the models you are training.
Parting thoughts
Hopefully the first part of the blog gives people who were not previously aware a taste of the large amount of work that many scientists and engineers have put into developing and understanding how models learn and how to build on that knowledge to solve tasks. These methods reflect years of refinement that enable their application at scale, as well as a degree of understanding that lets us break down their components and look at them piece by piece.
And the second part of the blog hopefully gives a taste for a field that is still in the initial phase, with rapid iteration on approaches, and at the beginnings for us to understand how best to use and apply. It will be exciting to see the developments that end up becoming essential in this new field. And if you think you know the best way, I’d be curious to read your blog on it.